Molecular Topology
NMRView applications are all about studying molecules, so it's not surprising that you can use it to keep track of information about your molecule. But how do you get all those bonds and atoms into the program where you can do something useful with them. Most NMRView users are studying molecules made up of amino acids and/or nucleic acids. Since these molecules are polymers of a small number of residues all you need to do is give NMRView a list of the names of the monomers. The simplest format for this information is just a text file containing the names of the monomers. Something like: met, ala, asn, glu, lys The entries in the file are the three letter names of the amino acids, and four letter names for nucleotides. For nucleotides, the first letter is "d" or "r" depending on whether the monomer is for DNA or RNA, respectively.
To read the file, use the menu command. If you like typing more than mousing, type the command "nv_sread seq fileName" and you will get the same result. In the current version of NMRView, the "fileName" argument needs to be the complete path to the molecule. Both these methods will read the molecule in, and setup up some information within NMRView so that when you save a "STAR" file the molecular structure will be saved as well. That way, you only need to explicitly read the molecule in once.
The "nv_sread seq" command allows you to read a structure without an external file by using the "-list" option. For example, nv_sread seq -list "ala gly phe" molname. Just be sure to have quotes or curly braces around the list of residues. In this example, "molname" specifies a name to be used for the molecule.
Lots of scientists are interested in studying the interactions of two molecules so you may want to have more than one molecule loaded at once. There is no hard limit in NMRView for the number of molecules. You'll probably have your head swimming in molecules before NMRView complains. To keep track of all these molecules they need to have a unique names..
By default the molecule name used within NMRViewJ is derived from the name of the sequence file. What if you want your file named myfavoritemoleculein2006.seq, but want to use a simpler name within the program, like Fred? Just put a name for the molecule in your sequence file. To do this you should include a molecule line at the beginning of the sequence file. This line should have two fields, the first should read "-molecule" and the second should be the name of the molecule. Two additional special lines can also be added to further define the molecule. The "polymer" and the "coordset" fields lines have the same format as the molecule line, that is "-polymer" or "-coordset" followed by the name. NMRViewJ will use them to allow multiple polymer and coordset entries. Think of the polymer as being a unique amino acid (or nucleic acid) sequence. The coordset corresponds to what X-ray crystallographers refer to as an assymetric unit. A homodimer would have one polymer, and two coordsets.
Ligands can be specified in the ".seq" file with a line like "-sdfile fileName.sdf". The file specified with the name must an ".mol" or ".sdf" file and must be in the same directory as the ".seq" file.
What if you already have a PDB file and don't want to be bothered writing a sequence file. Not too worry, you can read the PDB file directly with the menu command. You're probably wondering what the parenthetical "using library" comment is all about. If you use this command, then NMRView figures out what the residue sequence is from the PDB file, and then loads the appropriate residue topologies from NMRView's own residue library. The resulting atoms and bonds should be the same as if you had read a sequence file with the same sequence as the that in the PDB file. Because of this, the atom names may not be exactly the same as what's in the PDB file. If instead, you use the menu command, then NMRView will use the exact atoms that are in the PDB file. In this case NMRView figures out the bonding based on inter-atomic distances and it may not get the bonding exactly right.
You can also enter a molecular sequence directly within NMRVIEW without loading a file. Just use the menu item to display a sequence editor. You enter a molecule name at the top, and then enter a sequence using either the three letter or one letter codes. NMRVIEW will try to figure out which format you're using (space separated triplets of letters will be assumed to be the three-letter code, contiguous letters, like used in a Blast search will be assumed to be using the one letter codes, etc.).
The Topology from Sequence commands just generate molecular topology information within NMRView. (Whats topology? Just the names of the atoms, and what atoms are bonded to what other atoms.) There is no information about where in space (not to mention time) they are. You can have your topology and coordinates too (if you know what they are). If you've already generated a topology and want to add coordinates use the menu command. The above two methods that use PDB files will get coordinates for each atom they can find in the PDB file
When using the above menu option to read coordinates in NMRViewJ will read all the "models" in the PDB file. Each model will be stored in a structure accessible with the specified model number. For example, all the coordinates after "MODEL 4" will be in structure number "4" in NMRViewJ. All the models will be used when using the peak identification tools and when calculating atom rmsd values and molecular superpositions. When NMRViewJ first reads a sequence file it will automatically generate 3D coordinates for the atoms. These will be for the molecule in an extended conformation and will be stored in structure number "0". Structure number "0" is deselected in coordinates are explicitly read in. To specify which structures are currently active use the Structure Selector described below.
| Option | Entry |
|---|---|
| Simple sequence | met, ala, asn, glu, lys |
| Sequence starts at "5" | met 5, ala, asn, glu, lys |
| Sequence with breaks | met 5, ala, asn 8, glu, lys, thr, his 15, arg, thr |
| DNA/RNA sequence | dade, dthy, dgua, dcyt, rade, ruri, rgua, rcyt |
| Sequence with entity names | -molecule fred, -polymer chainA, -coordset mono1, met 5, ala, asn, glu, lys |
| Heterodimer | -molecule mymol, -polymer poly1, -coordset A, met, ala, asn, glu, -polymer poly2, -coordset A, val, asp, arg |
| Homodimer | -molecule mymol, -polymer poly1, -coordset A, -coordset B, met, ala, asn, glu |
| Polymer with ATP ligand | -molecule atpBinding, -polymer chainA, -coordset A, -sdfile atp.sdf, met, ala, asn, glu, lys |
| DNA with negative resnum | -molecule quad, -polymer A, -coordset A, da -2, dt, dg 1, dc, -polymer B, -coordset B, dg -2, dc da 1, dt |
Atom Nomenclature
Unfortunately, there are two schemes, rather than just one, for naming atoms that are in common use by NMR scientists. First, there is the IUPAC standard which was agreed upon by a committee of scientists who, we presume, hoped that their proposal would be universally adopted. Second, there is the nomenclature used by the program XPLOR. There are several differences between the nomenclatures, but the most obvious ones are in the naming of methylene protons, which in IUPAC are values like HB2 and HB3, or HG2 and HG3. In XPLOR these same atoms would be HB1 and HB2, and HG1 and HG2. Another notable difference is that of the amide proton, which in IUPAC nomenclature is "H", and in XPLOR is "HN". Occasionally, one will see a structure which uses IUPAC nomenclature for everything but the amide protons, which are labeled "HN" as with XPLOR. Any particular structure may also have deviations from either nomenclature, especially for the N-terminal protons and C-terminal oxygens. There seems to be no particular advantage for the continued use of the XPLOR nomenclature, other than inertia. Since it was already widely used at the time of the ratification of the IUPAC nomenclature it's continued use is, even if unfortunate, not surprising.
Some programs, for example CYANA, default to the use of the IUPAC nomenclature, and others, obviously XPLOR for one, default to the use of the XPLOR nomenclature. The consequence of this is a considerable loss of time and wasted effort in dealing with two nomenclatures. It is also a barrier to the facile use of multiple programs in one project.
NMRVIEW can support either nomenclature, and has some tools for inter-conversion. The primary factor in determining which nomenclature is in use is the choice of residue libraries that are used when a molecular topology is created. Two sets of libraries, reslib_iu for IUPAC nomenclature, and reslib for XPLOR nomenclature are available. The choice of residue library can be made in the Paths and Filenames section of the Preferences Dialog. Also, if you are creating a new project with the Project Browser you will be prompted as to whether to use IUPAC as the nomenclature for the project. Finally, when a STAR file is saved the current nomenclature setting will be saved in the STAR file. When the STAR file is reopened that setting will be used when reconstructing the molecular topology, regardless of what the reslib preference is currently set at.
If you are unsure what nomenclature your current molecule is using you can select the menu item. This will execute a procedure that examines the current atom names and will report which nomenclature most clearly matches. Sometimes one nomenclature will match best, but there will be some atoms which don't match. A selection of these will be reported. If the nomenclature is not IUPAC you will be given the option of changing the nomenclature to IUPAC. This will not only change the atom names in the molecular topology, but also the names of any atom labels for peaks that are interpretable.
The numbering of residues is a related issue. Some structures being studied correspond to a fragment of a larger macromolecule, or may have an N-terminal addition used to aid in the expression or stability of the molecule. In these cases the numbering may start at 1, corresponding to the actual residues in the structure, or start at some other number that represents the position of the first residue relative to the complete macromolecule. You can select menu item to have the option of adjusting the residue numbering. This will not only change the residue numbers in the molecular topology, but also the residue numbers of any atom labels for peaks that are interpretable.
Atom Specifiers
Within NMRViewJ, atoms can be specified using a syntax as follows:
coordSetName.entityName:residueNum.atomName
The meaning of residueNum and atomName are relatively obvious, though it should be noted that the residueNum can be prefixed with the single letter amino acid code of the specified residue. That is you could enter 32.CA or F32.CA for the CA atom of residue CA (which happens to be a phenylalanine). But what is a coordSet and entity? Imagine a dimer of two identical polypeptide chains. Each polypeptide chain as represented by an amino-acid sequence is, in the terminology of NMRViewJ, an "entity". In this example there is only one entity. However, the entity is represented twice (each monomer) in the actual molecule. Each of these monomers is, in the terminology of NMRViewJ, a "coordSet" (so named because in the molecular structure each monomer is represented by a set of coordinates). A molecule that is a heterodimer would have two entities, one for each polymer. Similarly, a protein with a bound ligand would also have two entities, one for the polymer and one for the ligand. Multiple coordsets and entities can coexist, so for example a homopolymer with a ligand on each polymer would have two entities, each represented once in each coordset.
Use the "mol select" command to select a set of atoms, then use "mol listatoms" to return a list of them, and use "foreach" to loop over them. Here's an example: This would print out all the chemical shifts of the protons in a molecule.
mol select atoms *:*.H*
foreach atom [mol listatoms] {
set ppm [nv_atom elem ppm $atom]
if {$ppm != ""} {
puts $ppm
}
}Note: the "nv_atom elem ppm" command now returns "" (an empty string) if the atom doesn't have an assigned chemical shift (which I think is a better design than returning some stupid large negative number).
Within NMRViewJ, atoms can be specified using a nomenclature as follows:
coordSetName:residueName.atomName
The meaning of residueName and atomName should be obvious, but what is a coordSet?
Imagine a dimer of two identical polypeptide chains. Each polypeptide chain as represented by an amino-acid sequence is, in the terminology of NMRViewJ, an "entity". In this example there is only one entity. However, the entity is represented twice (each monomer) in the actual molecule. Each of these monomers is, in the terminology of NMRViewJ, a "coordSet" (so named because in the molecular structure each monomer is represented by a set of coordinates).
Use the "mol select" command to select a set of atoms, then use "mol listatoms" to return a list of them, and use "foreach" to loop over them. Here's an example:
This would print out all the chemical shifts of the protons in a molecule.
mol select atoms \*:\*.H\* foreach atom [mol listatoms] {
set ppm [nv\_atom elem ppm \$atom] if {\$ppm != ""} { puts \$ppm } }Note: the "nv_atom elem ppm" command now returns "" (an empty string) if the atom doesn't have an assigned chemical shift (which I think is a better design than returning some stupid large negative number).
Structure Family Analysis
The structure tool displayed here can identify "core" regions of the structure, calculate superpositions using the specified residue and atom filters, and align all structures on a selected entry. In the calculate mode the "most representative" structure is identified (shown with an RMSD value of 0.0 in the table). RMSD values in the table are from each structure to that structure. The structure analysis tool is also used to activate and deactivate individual members of the family of structures. Deactivated ones will not be used in calculations involving, for example, NOE violations.
- Residues
A space separated list of residues which will be used in calculating superpositions of the structures. Hyphenated values indicate inclusive ranges (30-33, means residues 30,31,32 and 33). If the list is empty then all residues will be used. An appropriate list can be calculated with the "Core" tool (see below).
- Atoms
A comma separated list of atom names which will be used in calculating superpositions of the structures. If the list is empty then all residues will be used.
- Core
The atomic coordinates of different residues in a macromolecule are more or less well defined, depending both on molecular motions and on the number and precision of restraints used in there calculation. Well defined regions are referred to as the "core". Clicking this button will find the core regions of the current molecule. The resulting list of residues will be placed in the Residues section of this interface. NMRVIEW
Core identification is done by finding a representative model (see below), superimposing the remaining models on that one, and then calculating the atomic rmsd values on a per residue basis. Residues whose rmsd value is less than 2.0 times the median rmsd across all residues are considered core residues.
- Calc
Clicking this button will first find a representative model, and then calculate the rmsd of all models to that one. The RMSD values will be displayed in the RMS column of the table in this interface. The row with an RMS value of 0.0 corresponds to the representative model.
Calculation of the representative structure is done by doing a superposition of all possible pairs of models. The model whose average RMS deviation to the remaining models is lowest is the representative model.
- RMSD
Clicking this command will generate a plot of the residue RMSD values. The same calculation is used in the core identification tool mentioned above
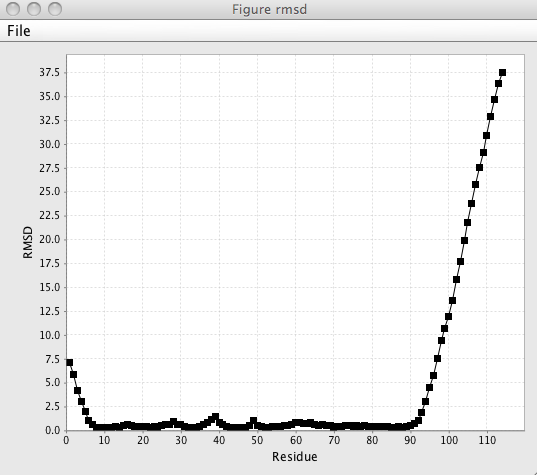
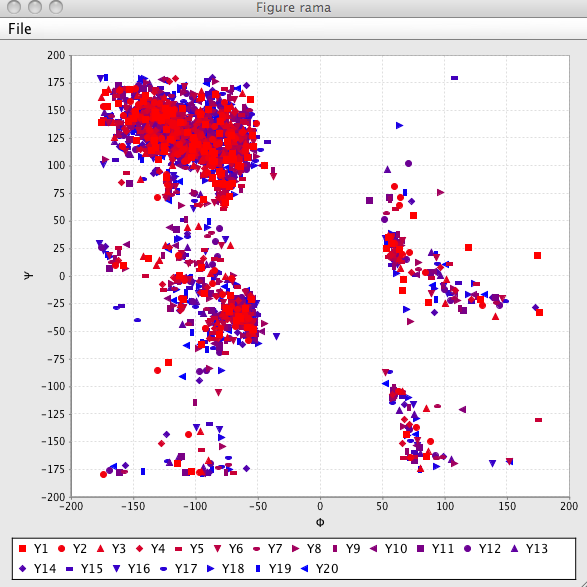
- Rama
Clicking the Rama button will generate a Ramachandran (Phi-Psi) plot of the structures. Only those structures that are activated (have a value in the state column of "1" will be used. Each model is displayed with a different color and symbol and a legend is displayed to show this symbol-model match.
The molecular viewer can display one or more structures to allow you to view the family of structures
RNA Viewer
This tool allows displaying the secondary structure of RNA and can include the display of distance constraints and deuterium labeling. Open the tool up by choosing the RNA Sec Structure menu item from the Molecule -> Analysis menu. If you're RNA molecule has not had a secondary structure assigned within NMRViewJ using the dot-bracket notation, then click the Dot-Bracket button. This will display the following dialog.
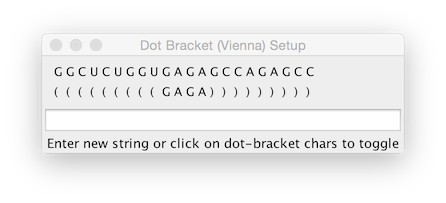
The dot-bracket notation is used to indicate base-pairing. A left parenthesis, is base paired to the corresponding base with a right parenthesis. Either a dot ("."), or the nucleotide base character can be used to indicate the base is not paired.
The first row of the display shows the sequence of RNA bases. The row below shows the dot-bracket assignment. By default this will consist of all "." values, indicating that there are no base pairs. You can update the assignment by typing dot-bracket notation into the text box, and hitting the Return key, or by clicking with the mouse on the assignment symbol below each base pair character. Clicking the mouse will toggle through values in the following order: "(",")","X","." (where X is the base pair character. If the number of left and right parentheses are not equal, the background will be displayed in yellow.
Clicking the Layout button will cause NMRViewJ to attempt to layout the sequence in a manner consistent with the base-pairing specified with the dot-bracket notation. This works well for relatively small RNAs. We have not yet optimized it for larger RNAs (which may take some time to layout). We plan to add the ability to import a file of x,y positions, so that the layout could be done in an external program.
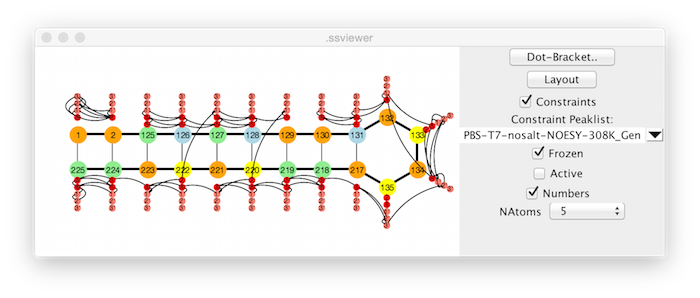
The display can be annotated with lines indicating the presence of distance constraints. If a Peaklist is not selected with the Constraint Peaklist selector, the constraints currently present in the NMRViewJ constraint table will be used. If a peak list is selected, then any assigned peaks in the peak list will be used (NOTE: this will overwrite any constraints currently present in the normal NMRViewJ constraints data).
If the Frozen checkbox is selected, then only peaks that have been marked as Frozen in the Peak Slider will be used for generating constraints. This allows a visual display of the location of frozen peaks. If the checkbox is not selected, then all peaks with assignments will be used.
If the Active checkbox is selected, then the display of hydrogen atoms (the smaller circles) will reflect the "active" state of atoms. The "active* state is normally used in the tool used to generate RNA Peaks (RNA Peak Generator). Atom types that are marked as deuterated (turned off in the Peak Generator) are inactive. Atom types marked as non-deuterated are active. Thus, the tool can be used to display the deuteration profile of the molecule.
If the Numbers checkbox is turned on, then the residue number rather than nucleotide base character (G,C,A,U) will be displayed.
The number of hydrogen atoms to be displayed can be specified with the NAtoms control. For example, a value of 0, turns all hydrogen atoms off, a value of 2 would display only the H2,H8,H5, and H6 (base) hydrogens. Larger numbers would additionally allow display of ribose hydrogens (in order H1', H2', H3', H4', and H5'). Constraint lines are only drawn to displayed hydrogens.
Template Library
When generating a molecular structure in NMRVIEW by reading in a sequence file it is necessary to translate the residue names into the set of atoms and bonds that define the molecular topology. To do this, NMRVIEW, looks at each residue name in the sequence file, and checks to see if a file corresponding to that name with a ".prf" extension exists in the residue library. If an appropriate residue file is not found in the standard directory (specified with the reslib entry in Preferences), then NMRVIEW can also look in a local residue library directory (specified with the local,reslib entry in Preferences). This is the appropriate place to place your own special .prf files. This way they won't be accidentally deleted when you upgrade to a new version of NMRVIEW.
If a residue file is found, that file is scanned to extract the topology for that residue. NMRVIEW works with residues using an internal coordinate system, where the molecular topology is determined by a tree-like structure starting from the first atom in the structure. To properly define the structure then, it is necessary to provide values for the valance angles, dihedral angles, bond distances and connectivity. Most of this information is defined in the ATOM and FAMILY records. The .prf files, which are derived from information originally published by Robson & Osguthorpe (J. Mol. Biol. 132:19-51) are described below. Our original use of .prf files was in our structure calculation program, PEGASUS (Johnson & Sugg, Biochem., 1992, 31,8151-8159). some of the parameters are only relevant to this structure calculation program, and do not need to be absolutely correct to use the .prf file for NMR assignment purposes.
LNAME Serine
SNAME ser
RCHAR S
ATOM N N' 1.32 114.00 -120.0
ATOM H H' 1.00 123.00 0.0
ATOM CA C1 1.47 123.00 180.0
ATOM HA H 1.08 109.47 -120.0
ATOM CB C2 1.53 109.47 -121.5
ATOM HB2 H 1.08 109.47 -120.0
ATOM HB3 H 1.08 109.47 -120.0
ATOM OG O 1.42 109.47 0.0
ATOM HG HO 1.00 110.00 0.0
ATOM C C' 1.53 109.47 120.0
ATOM O O' 1.24 121.00 180.0
FAMILY - N H CA
FAMILY N CA C CB HA
FAMILY CA CB OG HB3 HB2
FAMILY CB OG HG
FAMILY CA C + O
ANGLE CA PHI 1
ANGLE CB CHI1 2
ANGLE OG CHI2 6
ANGLE C PSI 1
ATREE CA CB C
ATREE CB OG
ATREE OG
ATREE C +
PSEUDO QB HB2 HB3
CRAD CA 4.0- LNAME
Full name of residue. Unused at present.
- SNAME
Short name of residue. Unused at present.
- RCHAR
Single letter name of residue. Unused at present.
- ATOM
Properties of the atoms in the residue. There should be one line for each atom. The line is composed of 6 fields separated by white space (spaces or tabs).
-
Atom name
-
Atom type, which should reference one of the atom types listed in the atoms.def file. Used for getting atomic number and energy parameters when doing structure calculations.
-
Bond length, from this atom to the previous atom in the tree structure of the residue (as defined in the FAMILY lines).
-
Valance Angle, between this atom, its parent, and grandparent, as defined in the tree structure of the molecule.
- Torsion Angle, between this atom, its parent, grandparent, and great grandparent (as defined in the tree structure of the molecule). The angle for the first "child" atom bonded to a given parent, as defined in the FAMILY lines, is an absolute torsion angle. The angles for subsequent atoms are relative to the previously defined dihedral.
-
- FAMILY
Defines the tree structure of the residue. Each line can be considered of the form
"FAMILY parentAtom thisAtom childAtom1 childAtom2...".
For example, a line like, "FAMILY N CA C CB HA", implies that the atom CA, is bonded to the N atom (the parent of CA is N), and it has three children, C, CB and HA. If the parent is specified as "-", then it is a connector atom in the previous residue. If a child is specified as "+", then it is the connector atom in the subsequent residue. Child atom names preceded with a "-", like-CD2 imply that this child atom will actually be defined in the tree structure in some other FAMILY entry in the structure, but that there should be a bond drawn between this child atom and the main atom of this FAMILY line. This is used to define bonds that close rings.
- ANGLE
Each rotatable bond in the residue has an ANGLE entry.
-
The rotatable bond is that between the specified atom and its parent.
-
The name of the angle (PHI, PSI, CHI etc.)
- A number indicating an entry in the file irp.def, which gives energy parameters for the intrinsic rotation potential for this atom. Only used in structure calculations.
-
- ATREE
One entry for each rotatable bond. The order in which they are specified gives the tree of rotation groups. Unused at present in NMRVIEW
- PSEUDO
Mapping of actual atoms in structure to pseudo atom names, "PSEUDO pseudoAtomName atom1 atom2 ...". The pseudo atom position would be at the geometric mean of all the actual atoms that are listed. At present this is only used when reading in constraint files using pseudo atom names (like CYANA .upl files) and is used to translate the pseudo atom name into the set of actual atoms stored in the molecular structure.
- CRAD
This specifies the name of an atom near the center of the residue and the approximate radius of a sphere around the central atom that would encompass all atoms of the residue. Used for accelerating calculation of non-bond contact list. Unused at present in NMRVIEW.