Molecular Viewer
The Crescent Molecular Viewer is, like all of NMRViewJ, a cross-platform tool written in the Java programming language. It makes use of the JavaFX 3D environment which gives access to hardware accelerated three dimensional graphics. The viewer is not designed to be a full featured molecular graphics program, but rather to be able to allow you to make effective use of molecular visualization while working on an NMR project. Because it is integrated directly inside NMRViewJ it is easy to use to visualize any internal 3D data. The viewer is implemented in a way that makes it analogous to other canvas windows in NMRViewJ. It can render various graphical items, from simple shapes like spheres and cylinders, to more complex items like full molecular structures. This allows us to use it to render molecules and associated information like constraints and the coordinate system of an orientation tensor. The Crescent Viewer is still under development, so features will be added and the interface changed in the coming versions.
The Crescent molecular viewer is a premium feature. That means it only runs if you have installed a current, valid license file. License files are available for users who are paying annual support. We don't like limiting availability of features in this way, but we need to have some financial support for NMRViewJ development, and this is one way we can encourage users to do so.
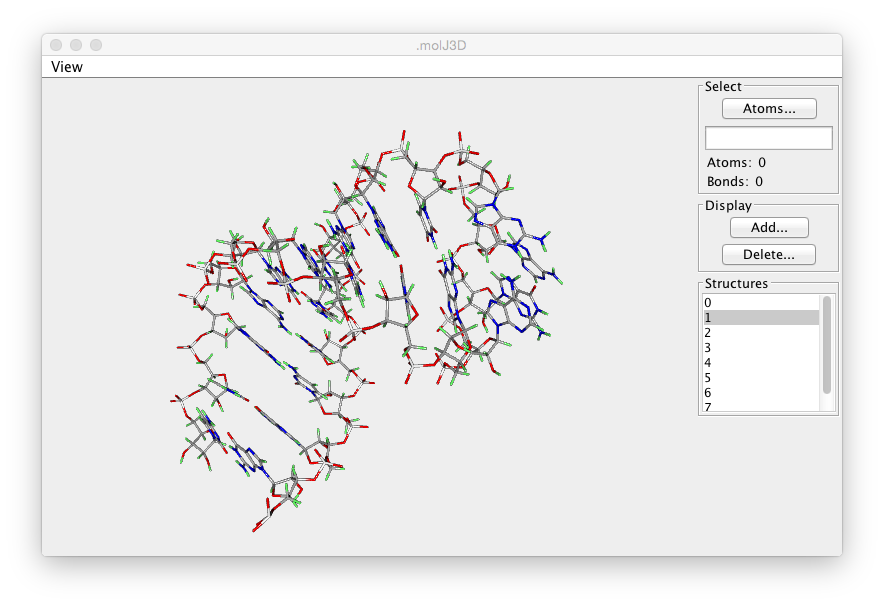
The Crescent Molecular Viewer will display the currently active molecule. See the previous chapter (Molecules) for information about loading molecular structures. Displaying the molecule consists of two steps, selecting the atoms to be displayed, and selecting the mode, lines or spheres, in which they are to be rendered. Molecules can also be rendered as a tube that follows the backbone and is colored with colors that sequentially change from one end to the other end of the molecule. When displaying a Tube, there is no need to first select atoms. A set of atoms appropriate to either a protein or nucleic acid will automatically be chosen when you click the Tube button.
The control sections at the right of the molecular viewer are described here.
- Select
Click Atoms... to select atoms based on one of the following choices: Backbone, Heavy, Sidechain, All, CAlpha, Active, and Current Residue. Choosing Active will select the atom (or atoms) that are currently selected in the molecular display. Choosing Current Residue will select all the atoms in residues that have an atom selected in the molecular display. Alternatively you can type in selections like 3.* to select all atoms in residue "3" or 5-10.N,C,CA to select backbone atoms in residues 5 through
- After entering a value, hit the Enter key to activate the selection. Whether you use the menu items, or explicit atom selections, the Atoms: and Bonds: fields will show the number of Atoms and Bonds that have been selected.
- Display
After selecting a set of atoms, click the Add button and choose from Lines, Sticks, Ball & Sticks, or Spheres button to render these atoms and bonds. Choosing one of these modes will add the atoms and bonds to the display list, and then clear the selection. You can repeat the cycle of Select-Display to display additional selections. You can also choose from Tube (to display a tube along the backbone), Box (to display a box that encloses all atoms), or Coordsystem (to display a coordinate system with x,y and z axes). Displaying Labels on atoms is currently under development.
- Delete
Clicking Lines, Spheres or Tube in the Delete selection will remove all of the corresponding objects from the display.
- Structures
If you have a whole family of structures loaded you can select which ones will be displayed using the list of structures at the bottom of the control region. Only structures highlighted in the list will be used in the display. If no structure is highlighted, then structure number 0 will be used. Remember that you can use the tools started from the menu item of the main menu.to align multiple structures.
The molecule can be rotated, translated or scaled as follows.
- Rotate
The molecular view can be rotated by pressing and then dragging with the left mouse button.
- Translate
The molecular view can be translated by pressing and then dragging with the middle mouse button..
- Scale
The molecular view can be scaled by pressing and then dragging with the right mouse button. Dragging right will increase the size of the viewed molecule, and dragging left will decrease it.
- Selection
Click with the left mouse button on atoms or bonds to select them. If the shift key is held down you will select an additional atom/bond without deselecting any existing ones.
The rotation, translation and scaling done with the mouse can be reset to the original transform by menu item on the molecular viewers menu bar. You can center the molecule by using the by menu item on the molecular viewers menu bar.